Create a free profile to get unlimited access to exclusive videos, sweepstakes, and more!
We finished sequencing the human genome, for real this time
What's a couple hundred million base pairs among friends?

The Human Genome Project officially launched on October 1, 1990, with the ambitious goal of sequencing the more than 3 billion base pairs that make up our DNA. The project took nearly 13 years, finally ending in April of 2003. The project was a huge success, having revealed the majority of our genetic makeup for the very first time. Yet, it was incomplete. Limitations in sequencing technology at the time meant that approximately 8% of the genome was unreadable.
Over the intervening years, that initial reference genome was improved, and some of the missing data was filled in, but errors and gaps remained. Our understanding of the human genome remained incomplete, until now.
More than a hundred scientists working together under the Telomere-to-Telomere Consortium, embarked upon a new sequencing project to deliver a complete human genome, filling in the missing pieces and correcting the errors present in the previous reference genomes. The successful completion of the project was announced in a paper published in the journal Science. We spoke with Mitchell Volger from the Department of Genome Sciences at the University of Washington School of Medicine about the project.
"In 2001, the technologies limited us so that we could only assemble about 92% of the genome, but with new technologies we can go after the whole genome. As our technology has gotten better, effectively the puzzle pieces have gotten bigger. That allows us to unambiguously put things together," Volger told SYFY WIRE.
The consortium, which takes its name from the protective caps of repeating non-coding DNA at the ends of chromosomes, took a surprising approach to map their genome. Rather than simply filling in the missing pieces and correcting the errors in one of the existing reference genomes, the T2T Consortium started from scratch, utilizing new sequencing technology to create a new reference genome called T2T-CHM13.
"In the original Human Genome Project, the genome that's represented is actually an amalgamation of many different human individuals. This new one is one biological genome, that's why we started from scratch," Volger said.
The second part of the name comes from the type of material they sequenced. Living people, or even standard embryos, contain genetic information from both parents, which makes isolating and identifying base pairs or groups of base pairs more difficult. To get around that challenge, the T2T team selected what’s known as a complete hydatidiform mole — or CHM — a fertilized egg in which the maternal chromosomes are missing and the paternal chromosomes are duplicated and two X chromosomes are present. Using a CHM simplifies the sequencing process by isolating a single, repeated, data set to look at. The only downside is there is no Y chromosome present.
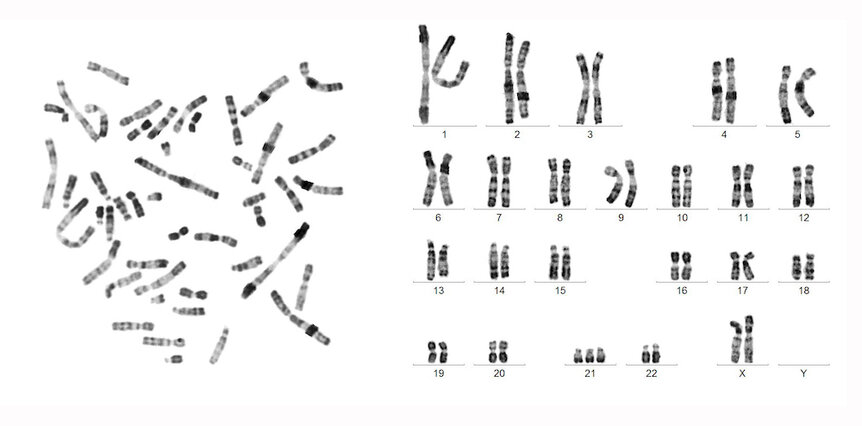
Advances in sequencing technology allowed the team to complete long-read sequences of a million letters of DNA at a time, orders of magnitude more than the previous reference sequences which had to read hundreds or thousands of letters at a time. As a result, they were able to more readily identify repeats and put the pieces back together correctly.
In the end, they were able to achieve a gapless sequence from telomere to telomere for all 22 human autosomes — the lettered chromosomes not related to biological sex — as well as the X chromosome. In total, they sequenced more than 3 billion base pairs.
"The sequencing for this project was done over the course of three to four years. The vast majority of the assembly we built in the end was automated. A lot of the time spent after that was making sure it was correct. I think going forward, this process will be much more routine," Volger said.
A majority of the information correlated to base pairs which were previously identified as part of the Human Genome Project or other reference genomes, but repeating the process for the known base pairs allowed them to correct errors which resulted from the less efficient and accurate technology available in the ‘90s and early 2000s. Moreover, roughly 182 million base pairs identified in the T2T-CHM13 genome didn’t correlate at all to previous references because they hadn’t previously been sequenced.
Among the genes uncovered are those responsible for our unusually large and connection-rich brains, as compared to our closest living relatives. They also added five full chromosome arms which were previously unmapped.
"There's a gene from a gene family called TBC1D3, it's a gene that exists multiple times in almost all humans. Chimpanzees only have one copy, but we have three or four or even more. If you express that gene in a mouse brain it induces cortical folding, which isn't normal in mouse brains. That gene family might be really important for how our brains developed," Volger said.
Mapping the Y chromosome using the same method is a near-term next step for the project, but they aren’t going to stop there. Scientists were clear that while this new sequence amounts to the most complete human genome ever, it does not capture the entirety of diversity present in our species. T2T is partnering with the Human Pangenome Reference Consortium to acquire a collection of 350 genomes across the range of human diversity in order to more fully catalogue what it is that makes humans human.
"One thing that's really exciting is that if we wanted to go and do this for another individual, we could probably generate the same resources in a month or two. We've laid a lot of groundwork that will allow us to go after a lot more genomes a lot more quickly," Volger said.
As with all scientific exploration and discovery, peering inside of ourselves is as much a work in progress as peering into the depths of space. With better tools and clearer vision, we’re writing a more complete and more exciting human story, one letter — or a million — at a time.


























